









contingut
Hemofília
Què es ?
L’hemofília és una malaltia hemorràgica genètica que impedeix la coagulació de la sang, provocant un sagnat inusualment llarg quan es lesiona i, de vegades, sense fer-lo. Es produeix per una mutació genètica que provoca la manca o absència de proteïnes de coagulació que impedeixen que un coàgul es formi prou fort per aturar el sagnat.
L’hemofília es considera una malaltia dels nens i afecta 1 de cada 5000 parts dels nens. No obstant això, les noies poden portar la mutació genètica i desenvolupar una forma menor de la malaltia. En la població total, 1 de cada 12 persones té hemofília. (000) Hi ha dos tipus predominants de malaltia: l'hemofília A i la B. La prevalença de l'hemofília A és superior a la de l'hemofília B (1/1 homes contra 6 000). (1).
L’hemofília era encara una malaltia molt debilitant i mortal fa unes dècades des de la infància i l’adolescència. Avui en dia, tractaments efectius però restrictius permeten frenar el sagnat i limitar els danys i discapacitats corporals en persones amb hemofília.
Símptomes
el nen. L'hemorràgia prolongada es produeix després d'una lesió o fins i tot d'un trauma lleu. Poden ser espontanis (per tant, intervenen en absència de traumes) en formes greus de la malaltia. El sagnat pot ser intern o extern. Tingueu en compte que el sagnat en una persona amb hemofília no és més intens, però que la seva durada és més llarga. Les hemorràgies als músculs (contusions) i a les articulacions (hemartrosi), principalment als turmells, genolls i malucs, poden provocar amb el temps rigidesa i deformitats invalidants, que poden provocar paràlisi.
La malaltia és encara més greu quan la quantitat de factors de coagulació a la sang és baixa (1):
- Forma greu: hemorràgies espontànies i freqüents (50% dels casos);
- Forma moderada: hemorràgies anormalment llargues després de ferides lleus i hemorràgies espontànies rares (del 10 al 20% dels casos);
- Forma menor: hemorràgies anormalment llargues però absència d’hemorràgies espontànies (del 30 al 40% dels casos).
Els orígens de la malaltia
La sang conté proteïnes, anomenades factors de coagulació, que permeten formar un coàgul sanguini i, per tant, aturar el sagnat. Les mutacions genètiques impedeixen la producció d’aquestes proteïnes. Si els símptomes associats a l’hemofília A i B són molt similars, aquests dos tipus de malaltia tenen un origen genètic diferent: l’hemofília A és causada per mutacions del gen F8 (Xq28) que codifiquen el factor VIII de coagulació i l’hemofília B per mutacions. en el gen F9 (Xq27) que codifica el factor IX de coagulació.
Els factors de risc
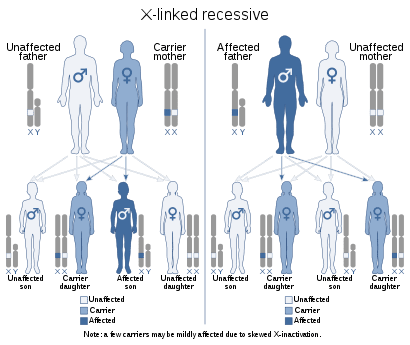
L’hemofília és causada per gens situats al cromosoma X. És una malaltia genètica coneguda com a "herència recessiva lligada a la X". Això implica que un home malalt transmetrà el gen alterat només a les seves filles, que el poden transmetre, amb un risc del 50%, a les seves filles i als seus fills. Com a resultat, la malaltia afecta gairebé exclusivament als homes, però les dones són portadores. Al voltant del 70% de l’hemofília té antecedents familiars. (1) (3)
Prevenció i tractament
Els tractaments ara permeten prevenir i frenar l'hemorràgia. Consisteixen en administrar per via intravenosa un factor antihemofílic: factor VIII per als hemofílics A i factor IX per als hemofílics B. Aquests fàrmacs antihemofílics es produeixen a partir de productes derivats de la sang (d'origen plasmàtic), o fabricats per enginyeria. genètica (recombinants). S'administren mitjançant injeccions regulars i sistemàtiques per prevenir l'hemorràgia o després d'un esdeveniment hemorràgic. La fisioteràpia permet a les persones amb hemofília mantenir la flexibilitat dels músculs i la mobilitat de les articulacions que han patit hemorràgies repetides.