









contingut
Síndrome de Treacher-Collins
Una síndrome genètica rara, la síndrome de Teacher-Collins es caracteritza pel desenvolupament de defectes congènits del crani i de la cara durant la vida embrionària, que es tradueixen en deformitats de la cara, les orelles i els ulls. Les conseqüències estètiques i funcionals són més o menys greus i alguns casos requereixen nombroses intervencions quirúrgiques. No obstant això, en la majoria dels casos, fer-se càrrec permet preservar una certa qualitat de vida.
Què és la síndrome de Treacher-Collins?
definició
La síndrome de Treacher-Collins (que porta el nom d’Edward Treacher Collins, que la va descriure per primera vegada el 1900) és una malaltia congènita rara que es manifesta des del naixement amb malformacions més o menys greus a la part inferior del cos. cara, ulls i orelles. Els atacs són bilaterals i simètrics.
Aquesta síndrome també s’anomena síndrome de Franceschetti-Klein o disostosi mandibulo-facial sense anomalies finals.
Causes
Fins ara se sap que hi ha tres gens implicats en aquesta síndrome:
- el gen TCOF1, situat al cromosoma 5,
- els gens POLR1C i POLR1D, situats als cromosomes 6 i 13 respectivament.
Aquests gens dirigeixen la producció de proteïnes que tenen un paper clau en el desenvolupament embrionari de les estructures facials. La seva alteració per mitjà de mutacions altera el desenvolupament d’estructures òssies (principalment les de les mandíbules inferiors i superiors i els pòmuls) i els teixits tous (músculs i pell) de la part inferior de la cara durant el segon mes de gestació. La pinna, el conducte auditiu i les estructures de l’orella mitjana (ossicles i / o timpans) també es veuen afectades.
diagnòstic
Es poden sospitar malformacions facials des de l’ecografia del segon trimestre de l’embaràs, especialment en casos de malformacions significatives de l’oïda. En aquest cas, el diagnòstic prenatal serà establert per un equip multidisciplinari a partir de la ressonància magnètica (RM) del fetus, que permetrà visualitzar les malformacions amb més precisió.
La majoria de les vegades, el diagnòstic es fa mitjançant un examen físic realitzat al néixer o poc després. A causa de la gran variabilitat de les malformacions, s’ha de confirmar en un centre especialitzat. Es pot ordenar una prova genètica en una mostra de sang per buscar les anomalies genètiques implicades.
Algunes formes lleus passen desapercebudes o es detectaran fortuïtament tard, per exemple després de l'aparició d'un nou cas a la família.
Un cop fet el diagnòstic, el nen se sotmet a una sèrie d’exàmens addicionals:
- imatges facials (radiografia, tomografia computada i ressonància magnètica),
- exàmens auditius i proves auditives,
- avaluació de la visió,
- cerca d'apnea del son (polisomnografia) ...
Les persones interessades
Es creu que la síndrome de Treacher-Collins afecta un de cada 50 nounats, tant en nenes com en nens. S'estima que cada any apareixen al voltant de 000 nous casos a França.
Els factors de risc
Es recomana assessorament genètic en un centre de referència per avaluar els riscos de transmissió genètica.
Al voltant del 60% dels casos apareixen aïllats: el nen és el primer pacient de la família. Les malformacions es produeixen després d’un accident genètic que va afectar una o altra de les cèl·lules reproductores implicades en la fecundació (mutació “de novo”). El gen mutat es transmetrà als seus descendents, però no hi ha cap risc especial per als seus germans. Tot i això, s’hauria de comprovar si un dels seus pares no pateix realment una forma menor de la síndrome i porta la mutació sense saber-ho.
En altres casos, la malaltia és hereditària. Molt sovint, el risc de transmissió és d’un de cada dos amb cada embaràs, però, segons les mutacions implicades, hi ha altres modes de transmissió.
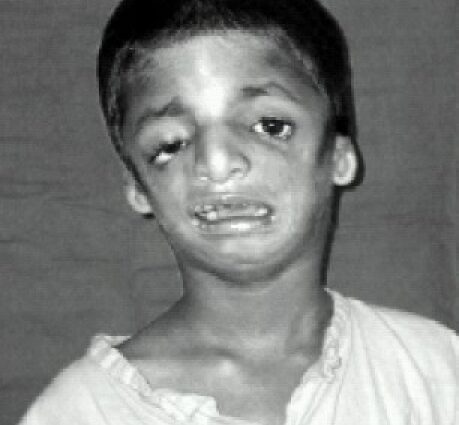
Símptomes de la síndrome de Treacher-Collins
Els trets facials dels afectats solen ser característics, amb una barbeta atrofiada i retrocedida, pòmuls inexistents, ulls inclinats cap avall cap a les temples, orelles amb pavelló petit i mal rodat, o fins i tot completament absents ...
Els principals símptomes estan relacionats amb malformacions de l’esfera ORL:
Dificultats respiratòries
Molts nens neixen amb vies respiratòries superiors estretes i boques estretament obertes, amb una petita cavitat oral en gran part obstruïda per la llengua. Per tant, tenen dificultats respiratòries importants, especialment en nadons i nadons, que s’expressen per roncs, apnea del son i respiració massa feble.
Dificultat per menjar
En lactants, la lactància materna es pot veure dificultada per dificultats respiratòries i per anomalies del paladar i del paladar tou, de vegades dividides. L’alimentació és més fàcil després de la introducció d’aliments sòlids, però la masticació pot ser difícil i els problemes dentals són habituals.
Sordesa
El malestar auditiu per malformacions de l’oïda externa o mitjana està present en un 30 a un 50% dels casos.
Alteracions visuals
Un terç dels nens pateixen estrabisme. Alguns també poden ser miops, hiperòpics o astigmàtics.
Dificultats d’aprenentatge i comunicació
La síndrome de Treacher-Collins no causa dèficit intel·lectual, però la sordesa, els problemes visuals, les dificultats de parla, les repercussions psicològiques de la malaltia i les alteracions induïdes per l’atenció mèdica sovint molt intensa poden provocar un retard. llenguatge i dificultats per comunicar-se.
Tractaments per a la síndrome de Treacher-Collins
Assistència infantil
Pot ser necessari suport respiratori i / o alimentació amb tubs per facilitar la respiració i l’alimentació del nadó, de vegades des del naixement. Quan s’ha de mantenir l’assistència respiratòria al llarg del temps, es realitza una traqueotomia (petita obertura a la tràquea, al coll) per introduir una cànula que garanteixi directament el pas de l’aire a les vies respiratòries.
Tractament quirúrgic de malformacions
Es poden proposar intervencions quirúrgiques més o menys complexes i nombroses, relacionades amb el paladar tou, les mandíbules, la barbeta, les orelles, les parpelles i el nas per facilitar l’alimentació, la respiració o l’oïda, però també per reduir l’impacte estètic de les malformacions.
Com a indicació, les escletxes del paladar suau es tanquen abans dels 6 mesos, els primers procediments cosmètics a les parpelles i els pòmuls a partir de 2 anys, l’allargament de la mandíbula (distracció mandibular) cap als 6 o 7 anys, recol·locació de la pinna de l’oïda als 8 anys, l’ampliació dels canals auditius i / o la cirurgia dels ossells d’uns 10 a 12 anys ... Es poden realitzar altres operacions de cirurgia estètica a l’adolescència ...
Audiòfon
De vegades, els audiòfons són possibles a partir dels 3 o 4 mesos quan la sordesa afecta les dues orelles. Hi ha diferents tipus de pròtesis disponibles segons la naturalesa del dany, amb una bona eficiència.
Seguiment mèdic i paramèdic
Per tal de limitar i prevenir la discapacitat, el control periòdic és multidisciplinari i convida a diversos especialistes:
- ORL (alt risc d’infecció)
- Oftalmòleg (correcció de trastorns visuals) i ortopista (rehabilitació ocular)
- Dentista i ortodoncista
- Logopeda…
Sovint és necessari un suport psicològic i educatiu.